How NVIDIA Turned 'Gaming GPUs Go Brrr' Into 'Actually We Can Read The Language of Life Now' (Part 2.1 of 2)
Welcome Back to the Future of Drug Discovery (Where Your RTX 4090 Has More in Common With Pfizer Than Fortnite)
In Part 1, we covered why protein folding is basically nature's most cursed optimization problem, how AlphaFold solved a 50-year-old grand challenge in biology, and why your body is running a protein manufacturing operation that would make Amazon's logistics look like a lemonade stand.
But AlphaFold had problems. It was slow. It only predicted structures, it didn't design new ones. And while it revolutionized structural biology, pharmaceutical companies wanted MORE.
- They wanted speed (screening thousands of candidates).
- They wanted design (custom proteins with specific functions).
- They wanted multimodal models (understanding sequence AND structure AND function).
- They wanted it yesterday.
This is where NVIDIA looked at the entire field of computational biology and basically said: "Hold my tensor cores." What followed was NVIDIA doing what NVIDIA does best, taking a computationally expensive problem, throwing GPUs at it, optimizing the absolute hell out of it, and then building an entire ecosystem around it that makes everyone else's solutions look like they're running on a TI-83 calculator.
Except this time, instead of making video games run faster, they're making drug discovery run faster. Your gaming GPU is now being used to design proteins that might cure cancer. Let that sink in while I explain how we got here.
Part 1: The Speed Problem (Or: Why "Hours Per Prediction" Is Unacceptable When You're Screening Drug Candidates)
AlphaFold Was Revolutionary But SLOW
AlphaFold 2 predictions took hours on a single GPU. For one protein. In academic research, this is absolutely fine. You predict a structure, publish a paper, get citations, maybe get tenure, everyone's happy. You're not in a rush because science moves at the speed of peer review, which is to say, glacially.
In drug discovery? This is a NIGHTMARE. Here's why:
Drug discovery requires SCREENING:
- You don't design one protein and hope it works
- You generate thousands of variants
- You test them computationally
- You synthesize only the best candidates
If each prediction takes hours, screening 10,000 variants takes... years. Pharmaceutical companies don't have years. They have quarterly earnings calls and investors asking why their billion-dollar R&D budget hasn't produced a drug yet.
They need SPEED.
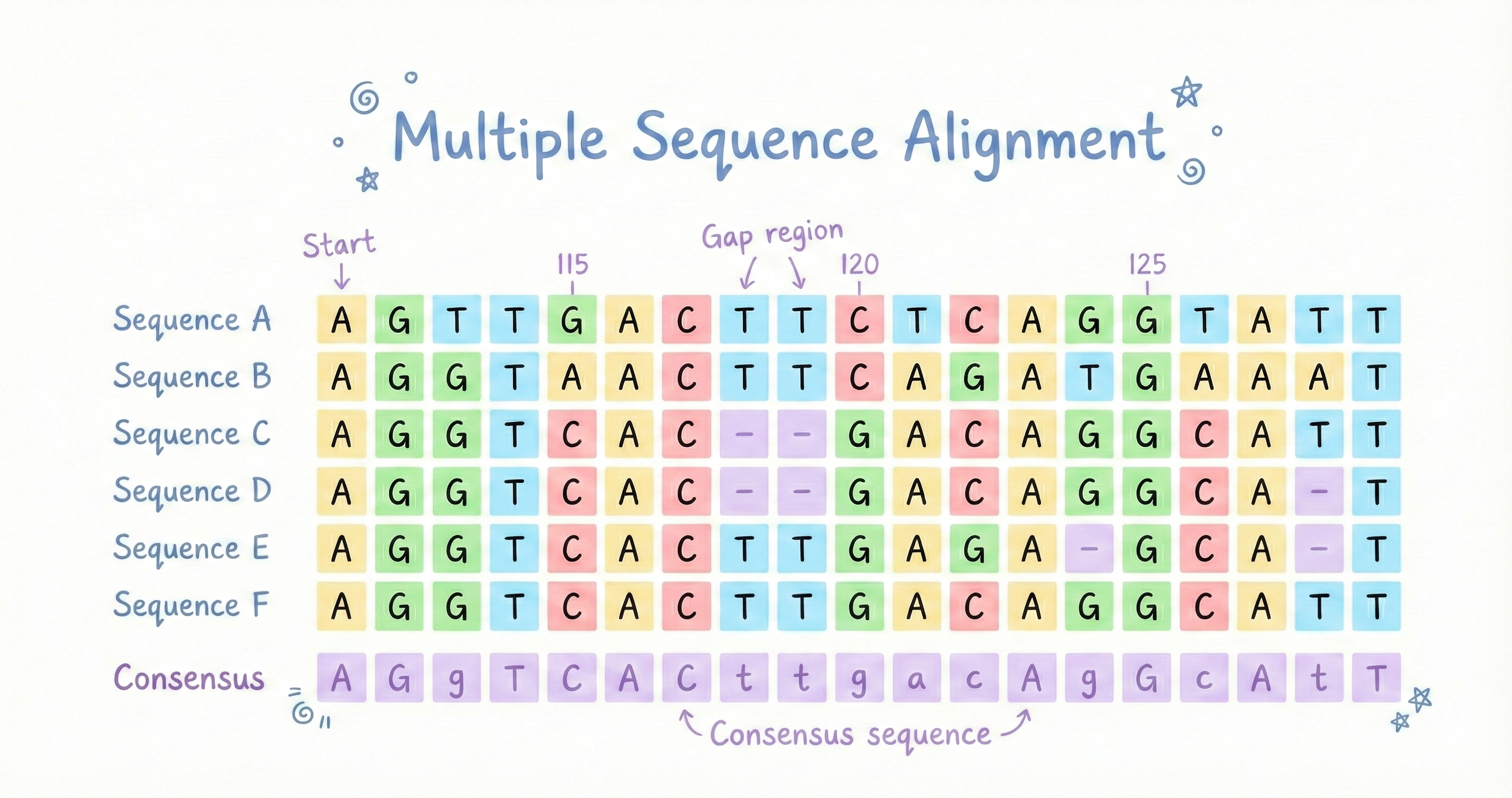
The Bottleneck: Multiple Sequence Alignment (MSA)
AlphaFold's workflow:
- Take a protein sequence
- Search databases for similar sequences (MSA generation) ← THIS IS SLOW
- Feed the MSA into a neural network
- Predict the 3D structure
The bottleneck? Step 2.
What is MSA (Multiple Sequence Alignment)?
MSA stands for Multiple Sequence Alignment, and it's basically evolutionary detective work. To predict how a protein folds, AlphaFold doesn't just look at your sequence in isolation. It looks at evolutionary information, how that protein evolved across different species over millions of years. Why? Because evolution conserves structure way more than sequence. Two proteins can have only 30% identical amino acid sequences but still fold into nearly identical shapes because the shape is what matters for function, and evolution keeps what works.
The MSA process:
- Search protein databases (hundreds of millions of sequences)
- Find proteins with similar sequences
- Align them to see which positions are conserved
- Use this to infer structural constraints (if two positions always mutate together, they're probably close in 3D space, coevolution signal)
AlphaFold uses tools like HHblits and JackHMMER for this searching and alignment work. The problem? These tools run on CPUs. And protein databases are absolutely massive, we're talking hundreds of millions of sequences. Searching through all that data on CPUs takes hours. Hours that pharmaceutical companies don't want to spend.
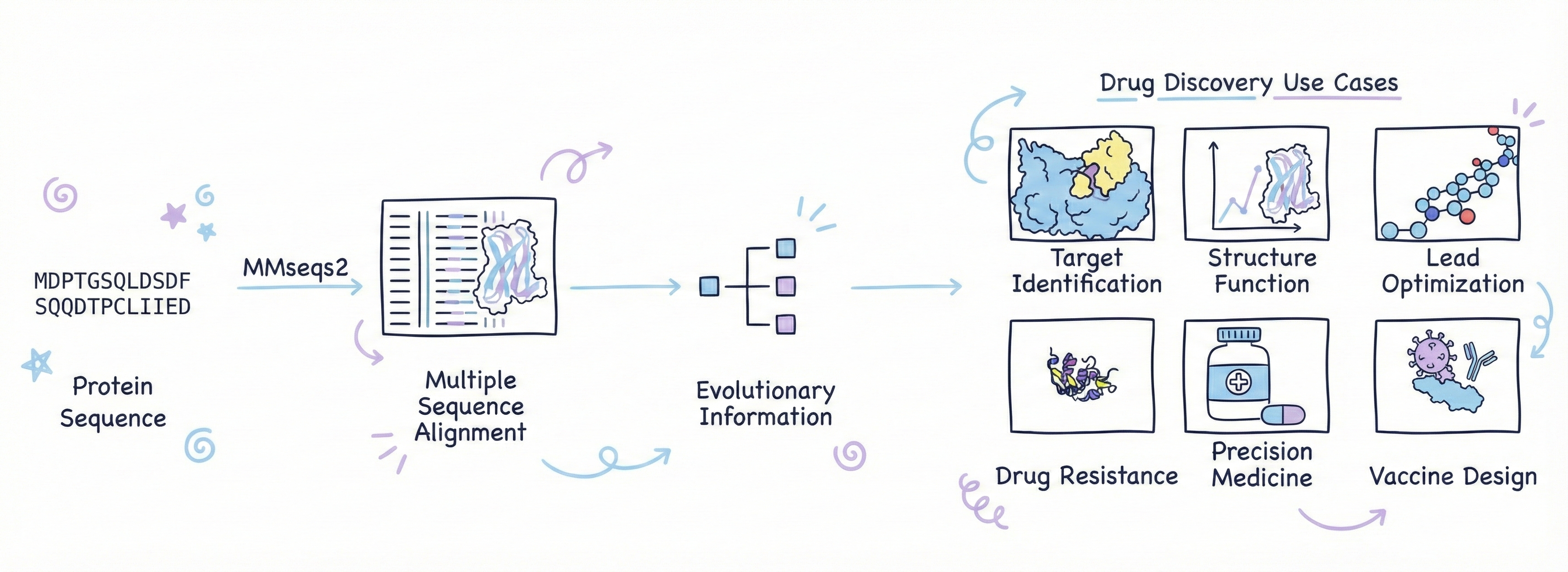
Enter: MMseqs2-GPU (The GPU-Accelerated Search Engine for Proteins)
NVIDIA looked at this situation and apparently said, "Why on earth are you using CPUs for what is obviously an embarrassingly parallel search problem?" They're not wrong. Searching databases for similar sequences is the kind of task that GPUs were literally designed for, massive parallelization of identical operations across different data.
So NVIDIA developed MMseqs2-GPU, which is exactly what it sounds like: a GPU-accelerated version of sequence alignment and database search. The performance gains are frankly absurd. On a single GPU, MMseqs2-GPU is 177 times faster than CPU-based methods. On eight GPUs working together, it's 720 times faster. Seven hundred and twenty times. The MSA bottleneck, previously the slowest, most painful part of the entire pipeline, became one of the fastest steps.
Why this matters?
Think about what this means in practical terms. The MSA search that used to take hours now takes minutes. For drug screening, this is the difference between "we can test 100 candidates this month" and "we can test 10,000 candidates this week." Speed equals more iterations, which equals better drugs, which equals faster time to market, which equals actually making money instead of burning through investor cash.
OpenFold: Making AlphaFold 138x Faster
AlphaFold 2 was originally implemented in JAX (Google's ML framework). It was designed for research, not production. OpenFold is a faithful PyTorch reproduction developed by the AlQuraishi Lab at Columbia. It:
- Matches AlphaFold's accuracy
- Is trainable (you can retrain it on custom data)
- Is optimized for NVIDIA GPUs
NVIDIA's optimizations:
- Custom CUDA kernels for critical operations (faster than PyTorch defaults)
- Non-blocking data pipelines (overlap data loading with computation)
- CUDA Graphs to reduce overhead
- Better multi-GPU parallelization
The result? AlphaFold inference with NVIDIA's optimizations runs 138 times faster than the original implementation. Let me repeat that: 138 times faster. What used to take an entire day now takes minutes. You can go from "I need to predict this structure" to "here's the structure" in the time it takes to get a coffee.
The training speedup is even more dramatic. The original AlphaFold 2 training took 11 days on 128 TPUs, which are Google's custom AI chips. OpenFold with NVIDIA's optimizations can train from scratch in 10 hours on 2,080 H100 GPUs. That's a 26-fold speedup in training time. Why does training time matter? Because pharmaceutical companies don't just want to use the pre-trained model, they want to retrain models on their own proprietary data to get better predictions for their specific use cases. Faster training means faster iteration on custom models.
Part 2: BioNeMo (Or: NVIDIA Trained GPT On 3.5 Billion Years Of Evolution Instead Of Twitter)
Here's the realization that fundamentally changed how we think about protein modeling, and honestly, it's so obvious in hindsight that you wonder why it took so long. Protein sequences are structured exactly like language. Think about it: an English sentence is a string of words drawn from a vocabulary of roughly 50,000 common words. A protein sequence is a string of amino acids drawn from a vocabulary of exactly 20 amino acids. Words in sentences follow grammar rules, syntax and semantics that determine meaning. Amino acids in proteins follow chemistry rules, thermodynamics and structural constraints that determine folding and function. Sentences convey meaning and information. Proteins convey structure and biological function.
If you can train an AI to understand language, and we absolutely can, that's what powers GPT, BERT, and every other large language model, then you can train an AI to understand proteins using the exact same techniques. This approach is called a protein language model, pLM, and it's become one of the most powerful tools in computational biology.

What BioNeMo Actually Is
BioNeMo is NVIDIA's framework and cloud platform for training and deploying large language models specifically for biology. Think of it as GPT for proteins, except it's not just proteins, it handles DNA, RNA, small molecules, basically any biological sequence data you can throw at it. It's a platform that provides pre-trained models you can fine-tune for specific tasks, a training framework optimized for biological data, and a cloud API so you don't need to own a supercomputer to use cutting-edge protein AI.
The genius of BioNeMo is that it packages up years of research into protein language models and makes them accessible. Instead of spending six months figuring out how to train a protein language model from scratch, you can use BioNeMo's pre-trained models and have predictions running in hours. Instead of buying millions of dollars worth of GPU infrastructure, you can hit a cloud API endpoint. It's democratizing access to technology that used to require being a major pharmaceutical company with a massive R&D budget.
BioNeMo includes multiple models:
ESM (Evolutionary Scale Modeling) Models
The cornerstone of BioNeMo is the ESM family of models, which were originally developed by Meta AI, yes, Facebook's research lab. Meta might be known for social media and questionable decisions about the metaverse, but their AI research is genuinely world-class. ESM-2, the current flagship model, has 3 billion parameters. For context, GPT-2, which was the original viral text generator everyone freaked out about, has 1.5 billion parameters. ESM-2 has double that, trained specifically on protein sequences.
How ESM works:
ESM uses the same transformer architecture as BERT. It's trained on millions of protein sequences using masked language modeling:
- Take a protein sequence:
MKTAYIAKQRQISFVKSHFSRQ... - Randomly mask some amino acids:
MKT[MASK]YIAKQR[MASK]ISFVKSHFSRQ... - Train the model to predict the masked amino acids based on context
- Repeat billions of times on millions of proteins
What ESM learns:
By training on sequences across all of evolution (bacteria, plants, animals, fungi), ESM learns:
- Which amino acids tend to appear together
- Which positions are conserved (important for structure/function)
- Which substitutions are tolerated (and which break the protein)
- Evolutionary patterns across billions of years
What ESM learns from this process is fascinating. By training on protein sequences from across all of evolution, bacteria, archaea, plants, animals, fungi, every domain of life, ESM learns fundamental patterns about proteins.
- It learns which amino acids tend to appear together because they work well structurally
- It learns which positions in proteins are highly conserved across species, which tells you those positions are critical for structure or function
- It learns which amino acid substitutions are tolerated, like you can swap leucine for isoleucine in certain positions without breaking the protein, and which substitutions are catastrophic.
- It learns evolutionary patterns that have been refined over billions of years.
When you feed a protein sequence into ESM-2, it outputs what's called an embedding, a numerical representation, essentially a long list of numbers that captures everything the model has learned about that protein. This embedding is incredibly useful for downstream tasks:
- Predicting whether mutations are harmful or benign (critical for understanding genetic diseases)
- Identifying functional sites like active sites in enzymes or binding pockets in receptors
- Predicting secondary structure (which parts form alpha helices vs beta sheets)
- Generating entirely new protein sequences
- Fine-tuning for any custom task you can imagine
Here's a real-world example that shows the power of this technology. Amgen, one of the world's largest biotechnology companies with annual revenue around $30 billion, took ESM-2 and fine-tuned it on their proprietary antibody data. These are sequences they've developed internally over decades of research, representing billions of dollars of investment. The results were dramatic: training custom models went from taking months to taking weeks. Inference, actually running predictions on new sequences, became 100 times faster than open-source tools they'd been using. Now they can predict antibody properties like binding affinity, stability, and immunogenicity, which is whether the antibody will trigger an immune response in patients. They're using this to design next-generation biologics, which are antibody-based drugs that represent the future of medicine for everything from cancer to autoimmune diseases.
MegaMolBART: Language Models for Chemistry
Proteins aren't the only molecules that matter in drug discovery. In fact, most drugs aren't proteins at all, they're small molecules, chemical compounds simple enough to manufacture at scale and stable enough to put in a pill. Think aspirin, ibuprofen, antibiotics, most of your medicine cabinet. These small molecules need AI tools too, and that's where MegaMolBART comes in.
MegaMolBART is a language model for chemistry, trained on 1.5 billion molecular structures from the ZINC database, which is basically the Library of Congress for drug-like molecules. The clever part is how molecules get represented as text.

How it works:
There's a format called SMILES, Simplified Molecular Input Line Entry System, that encodes molecular structures as strings of characters. For example, caffeine: CN1C=NC2=C1C(=O)N(C(=O)N2C)C
MegaMolBART reads SMILES strings exactly like GPT reads sentences. And once it understands the language of chemistry, it can do remarkable things. It can generate entirely new molecules with desired properties, maybe you want a molecule that's highly soluble in water for an injectable drug, or highly fat-soluble for a pill that needs to cross the blood-brain barrier. It can predict molecular properties like toxicity, which is critical because a drug that works great in a dish but kills liver cells is useless. It can figure out retrosynthetic pathways, which means starting with a target molecule and working backward to determine how to actually synthesize it in a lab using available chemicals and reactions.
That last capability is huge because chemistry is hard, and knowing whether a molecule can actually be made is as important as knowing whether it'll work as a drug. The impact on drug discovery is substantial. Traditional pharmaceutical research involves synthesizing thousands of molecules in the lab, testing each one individually, and watching 99% of them fail. It's expensive, time-consuming, and wasteful. With MegaMolBART, you generate candidate molecules computationally first. You predict their properties before anyone touches a beaker. You screen millions of candidates in silico, that's Latin for "in silicon," meaning on computers, and only synthesize the most promising ones. This cuts costs dramatically, accelerates timelines from years to months, and increases success rates because you're making educated predictions instead of educated guesses.
ProtT5: The Encoder-Decoder Model
ProtT5 is another protein language model in BioNeMo, but it uses an encoder-decoder architecture (like the original Transformer paper).
This means it can:
- Generate new protein sequences (decoder)
- Predict properties from sequences (encoder)
- Translate between different representations
Developed by the Rost Lab at Technical University of Munich.
Real application:
Startups like Evozyne are using ProtT5 (via BioNeMo) to design proteins for:
- Healthcare (treating congenital diseases)
- Clean energy (proteins that consume CO₂)
They've designed proteins that have never existed in nature and are testing them experimentally.

Why BioNeMo Beats Training Your Own Model
You might be thinking, "Okay, but can't I just download some code from GitHub and train my own protein language model?" And technically, yes, you could. Practically speaking, you absolutely should not, and here's why.
Training a 3-billion-parameter model like ESM-2 from scratch requires hundreds of GPUs running continuously for weeks. You need massive datasets, in this case, millions of protein sequences carefully curated from databases covering all of known life. You need expertise in distributed training, because getting hundreds of GPUs to work together efficiently is genuinely difficult and most machine learning engineers have never done it. You need to debug weird issues like gradient explosions, training instabilities, and optimization problems that only appear at scale. And you need to pay for all this. The compute costs alone easily exceed $100,000, probably closer to several hundred thousand if you're renting cloud GPUs. That's before we count engineering time, which for a team of experienced ML engineers over several months is another several hundred thousand dollars.
BioNeMo gives you pre-trained models for free. You can access them right now, no payment required. The catch, of course, is that if you want to do serious commercial work at scale, NVIDIA would very much like you to use their cloud infrastructure or buy their GPUs, but the models themselves are freely available.
Even if you have the budget to train from scratch, fine-tuning is dramatically cheaper and often works better. Instead of training on millions of generic proteins, you fine-tune a pre-trained model on your specific dataset, maybe antibodies from your company's proprietary library, or enzymes that work in industrial conditions, or membrane proteins for a specific therapeutic target. Fine-tuning takes days instead of months, costs thousands instead of hundreds of thousands, and often gives better results because the model starts with general protein knowledge and then specializes.
NVIDIA's optimization work means BioNeMo models are up to 100 times faster to train than naive open-source implementations. They use less memory, which means you can train bigger models or use larger batch sizes for faster convergence. They scale efficiently across multiple GPUs, so adding more hardware actually gives you proportional speedups instead of hitting diminishing returns. And crucially, they're designed to run on NVIDIA GPUs specifically, taking advantage of every architectural feature and optimization that NVIDIA has spent decades developing. The cloud API is perhaps the most democratizing aspect. You can access BioNeMo models with a simple HTTP request. No need to buy $50,000 worth of GPUs. No need to hire ML infrastructure engineers to set up your training pipeline. No need to maintain servers or deal with CUDA installation hell. You send a sequence, you get predictions back. It's not quite as simple as using ChatGPT, but it's close, and for a technology this powerful, that's remarkable.
Part 3: ProteinDT (Or: "Design Me A Protein That Does X" Actually Works Now)
Text-to-Protein Is Real
Remember when I said proteins are like language? NVIDIA took that idea completely literally and built ProteinDT, Protein Design with Text. This is a system that lets you design proteins by describing what you want in plain English. Not kidding. Not exaggerating. Actually plain English.
Here's an example of how it works:
- You type: "Design a protein that binds to EGFR receptor with high affinity." EGFR is Epidermal Growth Factor Receptor, a protein that's overactive in many cancers.
- You hit enter. The model thinks for a moment.
- And it outputs a protein sequence that, when folded, should have a binding pocket geometrically shaped to fit EGFR like a key in a lock.
That protein sequence can be synthesized in a lab, expressed in cells, purified, and tested to see if it actually binds to EGFR.
This feels like science fiction. It feels like the computer interfaces in movies where you just tell the computer what you want and it does it. Except it's real, it works, and companies are using it right now to design drug candidates.
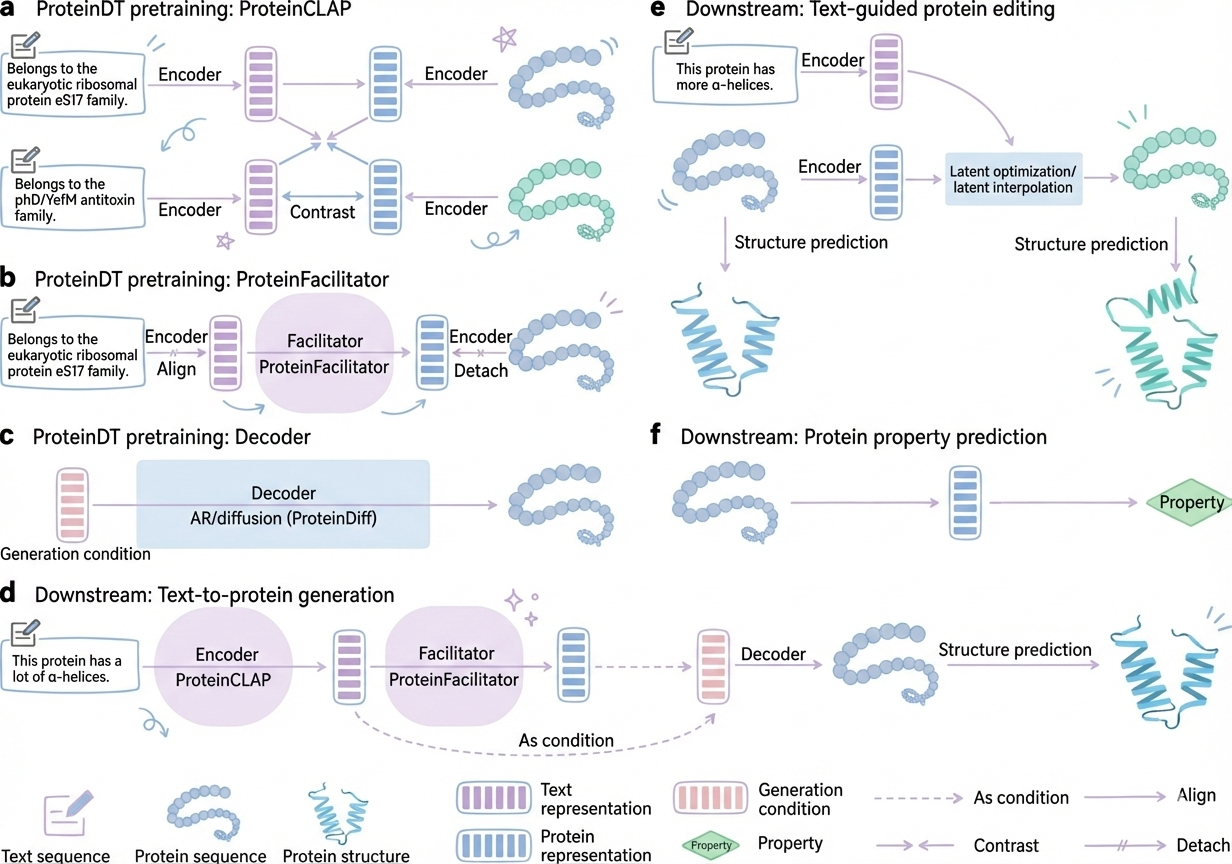
How ProteinDT Works
ProteinDT is what's called a multimodal model, meaning it works with multiple types of data simultaneously. Specifically, it connects two very different modalities: natural language text descriptions and protein sequences and structures. The model is trained to learn the relationship between how we describe proteins in words and what those proteins actually look like at the molecular level.
The training data for this is fascinating. You need millions of protein sequences, obviously. But you also need functional annotations for all those proteins, descriptions of what they do, where they're located in cells, what they bind to, what reactions they catalyze. These annotations come from decades of biological research carefully curated into databases like UniProt and Gene Ontology. You need text descriptions from scientific literature, millions of papers describing protein functions. And you need experimental data linking specific sequences to specific functions, like binding affinity measurements or enzyme kinetics.
It's trained to learn the relationship between language and protein properties.
The training data:
- Millions of protein sequences
- Functional annotations (what each protein does)
- Text descriptions from scientific literature
- Experimental data (binding affinities, enzyme kinetics, etc.)
What it learns:
ProteinDT learns that:
- "Binds DNA" → zinc finger motif (specific structural pattern)
- "Catalyzes ester hydrolysis" → serine protease active site
- "Recognizes phosphotyrosine" → SH2 domain
These functional motifs are structural patterns that perform specific functions. ProteinDT learns which sequences fold into which motifs.
Why This Works: The Biological Basis
The reason ProteinDT can work at all is because proteins have modular architectures. They're not just random sequences, they're built from functional motifs that can be mixed and matched like LEGO bricks. This is called domain architecture in biology, and it's fundamental to how proteins evolved.
For example, zinc finger motifs bind DNA with a very specific pattern: cysteine, then two variable amino acids, another cysteine, then twelve variable amino acids, then histidine, then three variable amino acids, then another histidine, so the specific pattern becomes Cys-X₂-Cys-X₁₂-His-X₃-His. Those four residues, two cysteines and two histidines, coordinate a zinc ion in the center, and the loop of amino acids between them forms a structure that slots into the major groove of DNA. This motif is found in thousands of different proteins, often repeated multiple times in the same protein to bind multiple DNA sites.
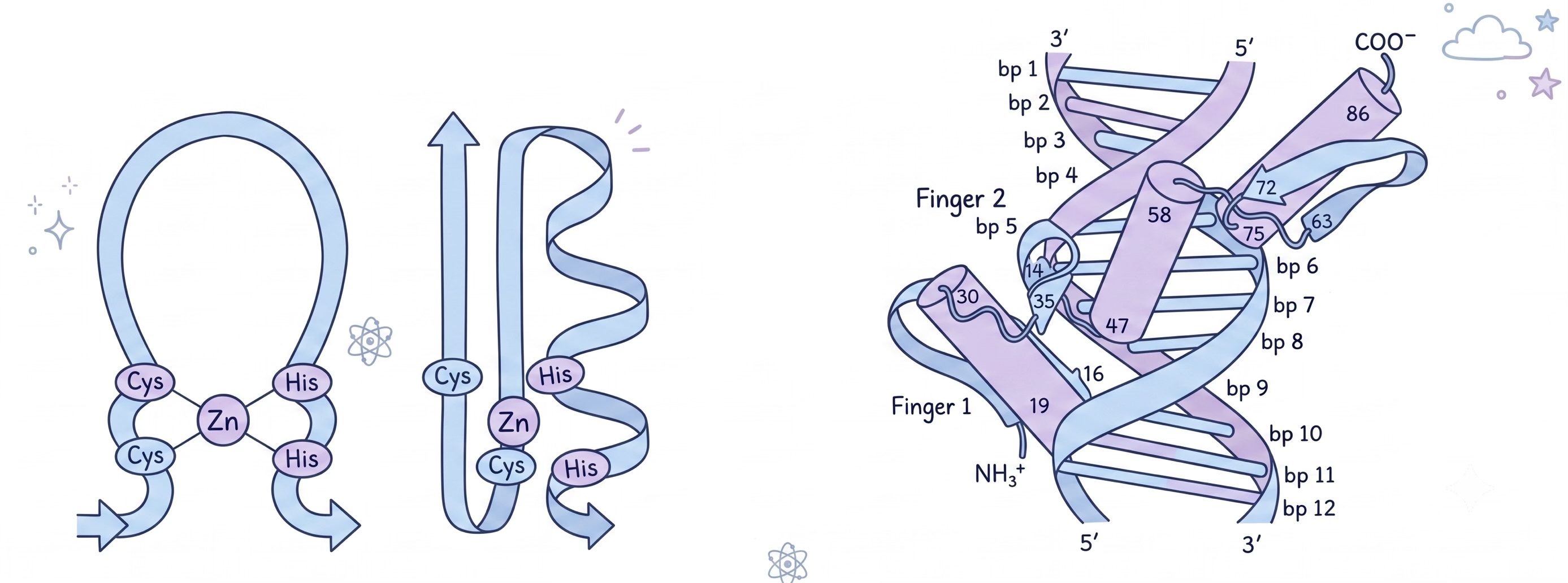
SH2 domains recognize phosphorylated tyrosine, which is a signaling modification cells use to activate proteins. The SH2 domain is about 100 amino acids with a deep binding pocket. The pocket has a positively charged patch that attracts the negatively charged phosphate group, and a hydrophobic region that accommodates the tyrosine's aromatic ring. This domain appears in hundreds of signaling proteins, always doing the same job: find phosphorylated tyrosines and bind to them.
Enzyme active sites have specific geometric arrangements of amino acids that catalyze reactions. Serine proteases, which cut peptide bonds in proteins, have a catalytic triad: serine, histidine, and aspartate arranged in a specific 3D configuration. The serine does the actual cutting, the histidine shuttles protons around to make the reaction work, and the aspartate stabilizes the histidine. This exact arrangement is found in digestive enzymes, blood clotting factors, and immune proteases.
ProteinDT learns all these patterns from data. It learns which sequences fold into which motifs, which motifs perform which functions, and how to combine them into complete, functional proteins. When you ask it for a protein that does something specific, it's composing functional motifs it's learned from millions of years of evolution.
Why This Is Revolutionary
Traditional protein design:
- Decide what function you want
- Figure out what structure would have that function (requires expert knowledge)
- Design a sequence that folds into that structure (requires computational tools + trial and error)
- Synthesize and test experimentally (months in the lab)
With ProteinDT:
- Describe what you want
- Get sequences
- Test
You've collapsed steps 2 and 3 into "ask the AI."
Real applications:
- Custom enzymes: "Design an enzyme that breaks down PET plastic" → generates candidates
- Therapeutic proteins: "Design an antibody that binds to this cancer marker" → generates antibody sequences
- Biosensors: "Design a protein that fluoresces when it binds glucose" → generates sensor proteins
Companies are using this. Right now.